Zespół Smitha Lemli Opitza to wrodzone zaburzenie rozwojowe charakteryzujące się między innymi charakterystycznymi rysami twarzy, niepełnosprawnością intelektualną i trudnościami w uczeniu się, problemami behawioralnymi oraz małą głową (małogłowie). Oprócz wad rozwojowych ważnych narządów, takich jak nerki, serce, narządy płciowe i przewód pokarmowy, dzieci z tym schorzeniem wykazują cechy autyzmu i zespołu nadpobudliwości psychoruchowej z deficytem uwagi (ADHD).Większość osób z tą chorobą ma zrośnięte drugie i trzecie palce, a niektórzy mogą mieć dodatkowe palce. Stan ten występuje stosunkowo rzadko i dotyka około jednego na 20 000 do 60 000 niemowląt.
SeventyFour / Getty ImagesObjawy
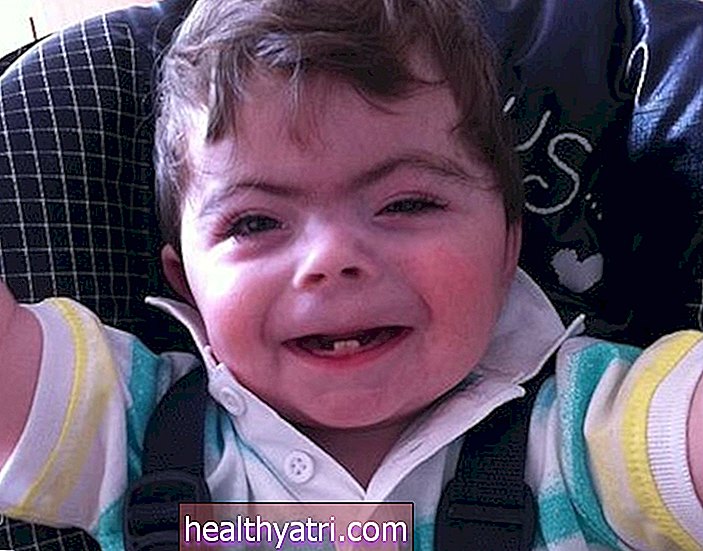
Objawy zespołu Smitha Lemli Opitza są obecne przy urodzeniu, a ich nasilenie jest bardzo zróżnicowane. W 80–99% tych przypadków. te cechy są widoczne:
- Palce z płetwami: Wspólną cechą tego schorzenia jest stapianie się drugiego i trzeciego palca, stan zwany „syndaktylem”.
- Niepełnosprawność intelektualna: chociaż stopień tego może się różnić, stan ten często prowadzi do trudności w uczeniu się.
- Nienormalnie mała czaszka: Mniejszy niż przeciętny rozmiar czaszki, stan zwany małogłowiem, to kolejna cecha charakterystyczna.
- Nieprawidłowe rysy twarzy: osoby z zespołem Smith Lemli Opitz mają charakterystyczne rysy twarzy, w tym mniejszą dolną szczękę i szeroki, płaski nos. W rzadszych przypadkach osoby mogą mieć opadające powieki, oczy kota, małe lub nieobecne oczy, a także szerokie usta.
- Trudności w karmieniu: u niemowląt stan ten może prowadzić do trudności w karmieniu piersią, wpływając na rozwój.
- Niższe napięcie mięśniowe: Wspólną cechą tego zespołu jest niższe niż przeciętne napięcie mięśniowe.
Istnieje wiele rzadszych objawów, występujących w dowolnym miejscu od 5% do 79% przypadków, w tym:
- Nieprawidłowości w rozwoju zębów: Wczesne wyrzynanie się zębów dorosłych i powiększone dziąsła są objawami zespołu Smitha Lemli Opitza.
- Niejednoznaczne genitalia: genitalia osób dotkniętych chorobą mogą być mniej zdefiniowane. Mężczyźni są bardziej narażeni na to, z niedorozwiniętym penisem i niezstąpionymi jąderami.
- Zespół nadpobudliwości psychoruchowej z deficytem uwagi (ADHD): To zaburzenie rozwojowe charakteryzuje się trudnościami w regulacji zachowania i impulsów, a także nadpobudliwością.
- Autyzm: Znany również jako zaburzenie ze spektrum autyzmu (ASD), stan ten prowadzi do upośledzonych umiejętności społecznych, mowy i zdolności komunikacji niewerbalnej, a także powtarzalnych zachowań.
- Wady serca: Wady serca związane z zespołem Smitha Lemli Opitza obejmują powstanie dziury w ścianie między dwiema górnymi komorami (ubytek przegrody międzyprzedsionkowej) lub jednej między dolnymi jamami (ubytek przegrody międzykomorowej).
- Zmieniona anatomia dłoni: osoby z tą chorobą mogą mieć dodatkowe małe palce u rąk i nóg. Ponadto położenie kciuka może być nietypowe, ponieważ znajduje się bliżej nadgarstka. Zgłaszano również palce błoniaste. Zgłaszano również pazury, nietypowe skrzywienie palców.
- Wrażliwość na światło: W wielu przypadkach skóra osób dotkniętych chorobą jest wyjątkowo wrażliwa na światło słoneczne.
- Częste infekcje: osoby z zespołem są narażone na zwiększone ryzyko infekcji bakteryjnej.
- Rozszczep języka: w około pięciu do 30 procent przypadków osoby dotknięte chorobą będą miały rozszczep języka, w którym końcówka jest podzielona.
- Nieprawidłowości w kręgosłupie: Wraz z innymi deformacjami kręgów, schorzeniu może towarzyszyć skolioza - boczna skrzywienie kręgosłupa - jak również kifoza lub garbus.
- Napady padaczkowe: osoby z tym stanem są bardziej podatne na napady padaczkowe.
- Mimowolne ruchy oczu: niekontrolowane i szybkie ruchy oczu (oczopląs) mogą również towarzyszyć zespołowi.
Przyczyny
Zespół Smitha Lemli Opitza to zaburzenie genetyczne spowodowane mutacją genu DHCR7. Ten gen reguluje ważny enzym, reduktazę 7-dehydrocholesterolową, która bierze udział w produkcji cholesterolu w organizmie. Wśród swoich funkcji cholesterol jest głównym składnikiem błon komórkowych i pomaga w tworzeniu mieliny, substancji chroniącej komórki mózgowe (neurony). Odgrywa również znaczącą rolę w prawidłowym trawieniu.
Mutacja DHCR7 powoduje brak reduktazy 7-dehydrocholesterolu powodując deficyty w produkcji cholesterolu. Pozwala również na gromadzenie się toksycznych produktów ubocznych cholesterolu w organizmie, co hamuje rozwój i wzrost wielu układów organizmu. Dokładny mechanizm tego, w jaki sposób ten brak cholesterolu prowadzi do zespołu Smitha Lemli Opitza, wciąż jest badany.
Defekt genetyczny, ten stan przebiega zgodnie z tak zwanym „autosomalnym recesywnym wzorcem”, co oznacza, że obie kopie genu - po jednej od każdego z rodziców - są niezbędne do jego rozwoju. Oznacza to, że rodzice osób z tą chorobą są nosicielami tego genu, ale sami niekoniecznie mają objawy.
Diagnoza
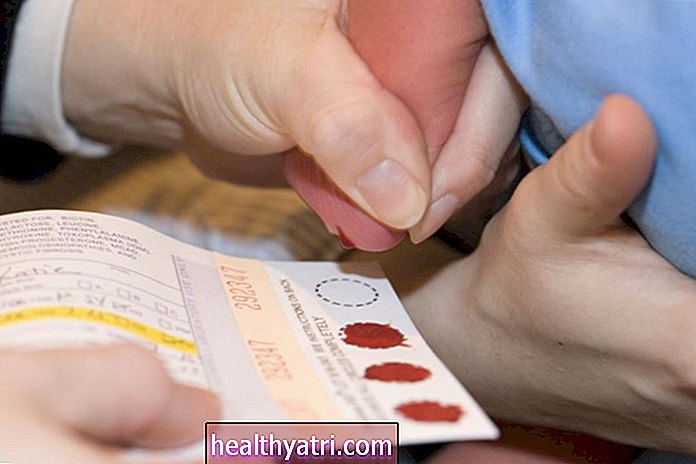
Podobnie jak w przypadku innych chorób wrodzonych, rozpoznanie Smitha Lemli Opitza obejmuje ocenę objawów fizycznych oraz badanie stosunku reduktazy 7-dehydrocholesterolowej do cholesterolu na podstawie badań krwi podejrzanych przypadków. Ponadto prenatalne testy genetyczne mogą również wykryć mutacje genu DHCR7, które prowadzą do rozwoju choroby.
Leczenie
Przyjmowanie tego warunku wymaga skoordynowanego wysiłku; ponieważ nie ma bezpośredniego lekarstwa na ten stan, należy skutecznie leczyć objawy i objawy. Takie podejścia obejmują:
- Suplementacja cholesterolu: chociaż potrzeba więcej badań, aby ocenić skuteczność tego podejścia, dieta bogata w cholesterol - obok przyjmowania suplementów - może pomóc złagodzić niektóre objawy.
- Fizjoterapia: Podejścia fizjoterapii i terapii zajęciowej, gdy są dostarczane w odpowiednim czasie, mogą pomóc w przypadku niepełnosprawności związanej z chorobą.
- Leczenie: Dostępne są metody leczenia niektórych fizycznych objawów zespołu Smitha Lemli Opitza, w tym problemów trawiennych, problemów wzrokowych, a także deformacji twarzy i innych.
- Nadzór: Skuteczne zarządzanie tym stanem wymaga konsekwentnego monitorowania objawów fizycznych, opóźnień rozwojowych i czynników dietetycznych.
Rokowanie
Dobra wiadomość jest taka, że jeśli zespół Smitha Lemli Opitza jest właściwie zarządzany i zapewnia się odpowiednią opiekę medyczną, osoby z tą chorobą mogą mieć normalną długość życia. To powiedziawszy, niezależne życie jest mało prawdopodobne ze względu na poważną niepełnosprawność intelektualną, często towarzyszy temu zespołowi. Warto zauważyć, że przeżywalność niemowląt z poważnymi objawami jest poważnie ograniczona i istnieje ryzyko śmierci w ciągu kilku miesięcy.
Korona
Poważne zaburzenie wrodzone, takie jak zespół Smitha Lemli Opitza, stanowi poważne wyzwanie zarówno dla osoby dotkniętej chorobą, jej rodziny, jak i lekarzy. Mimo że skuteczne zarządzanie jest możliwe, nie ma wątpliwości, że ten ciężar ma poważne konsekwencje psychologiczne. Osobom, które zajmują się opieką nad osobą z tą chorobą, pomocne mogą okazać się poradnictwo lub grupy wsparcia dla osób niepełnosprawnych. W szczególności zasoby, takie jak linki do najnowszych badań i usług wsparcia, są gromadzone razem przez Smith Lemli Opitz / RSH Foundation.
Słowo od Verywell
Stan, który jest tak wyniszczający i trudny, który może wpływać na tak wiele aspektów jakości życia, może wydawać się przytłaczający. To powiedziawszy, nie tylko istniejące metody leczenia zespołu Smitha Lemli Opitza są stale udoskonalane i ulepszane, ale trwają badania nad tym zaburzeniem. Gdy społeczność medyczna dowie się więcej o przyczynach i skutkach tego schorzenia - a także o skuteczności metod leczenia - rokowanie i jakość życia osób dotkniętych chorobą tylko się poprawią.