Zanik mięśni kręgosłupa (SMA) jest rzadką chorobą genetyczną wpływającą na nerwy kontrolne rozgałęziające się od rdzenia kręgowego na mięśniach prążkowanych. SMA dotyka głównie dzieci.
Dziecko z SMA będzie doświadczać upośledzenia kluczowych funkcji, takich jak oddychanie, ssanie i połykanie. W wyniku takiego upośledzenia mogą powstać dodatkowe warunki. Na przykład, z powodu słabych mięśni pleców mogą rozwinąć się nieprawidłowe krzywe kręgosłupa, co dodatkowo komplikuje proces oddychania poprzez ucisk na płuca.
Przed pojawieniem się zgłębników, przerwy w połykaniu często powodowały śmierć w przypadku SMA typu 1 (najpoważniejsza postać). Obecnie istnieje wiele urządzeń wspomagających, które pomagają utrzymać dzieci z SMA przy życiu (i czują się komfortowo, przynajmniej w porównaniu z poprzednimi latami).
Jednak nadal istnieje ryzyko. Jeden się dusi. Zadławienie jest możliwe, ponieważ dziecko z SMA ma słabe mięśnie przełykania i żucia. Innym ryzykiem jest aspiracja lub wdychanie pokarmu. Aspiracja może zablokować drogi oddechowe, a także być źródłem infekcji.
SMA przejawia się na wiele sposobów, które będą się różnić, szczególnie w zależności od typu. We wszystkich typach SMA można spodziewać się osłabienia mięśni, wyniszczenia i atrofii, a także problemów z koordynacją mięśni. Przyczyna tego leży w naturze samej choroby - SMA wpływa na kontrolę nerwową mięśni dobrowolnych.
Nie ma lekarstwa na SMA. Najbardziej obiecujące rokowanie wiąże się z wczesnym wykryciem. Postępy medycyny mogą pomóc w radzeniu sobie z powikłaniami związanymi z SMA.
Rodzaje rdzeniowego zaniku mięśni
Rdzeniowy zanik mięśni dotyka 1 na 6000 noworodków. Jest to główna genetyczna przyczyna śmierci dzieci w wieku poniżej 2 lat. SMA nie dyskryminuje, kogo dotyka.
Istnieje kilka rodzajów SMA, w zależności od stopnia nieprawidłowości obserwowanych w przypadku białka SMN. Istnieją również odmiany SMA, które są związane z innymi problemami genetycznymi.
SMA dzieli się na kategorie według nasilenia i wieku wystąpienia objawów. Stopień ciężkości, ilość niedoboru białka w neuronach ruchowych i (wczesny) wiek zachorowania mają tendencję do wykazywania korelacji między sobą. Rozwój doznań i umysłu w SMA jest całkowicie normalny.
Typ 1
SMA typu 1 jest najcięższą, dotykającą dzieci w wieku poniżej 2 lat. Rozpoznanie SMA typu 1 zwykle stawia się w pierwszych sześciu miesiącach życia.
Niemowlęta z SMA typu 1 nigdy nie są w stanie osiągnąć normalnych osiągnięć w zakresie rozwoju motorycznego, takich jak ssanie, połykanie, przewracanie się, siedzenie i raczkowanie. Dzieci z SMA typu 1 zwykle umierają przed ukończeniem 2 roku życia, zwykle z powodu problemów z oddychaniem.
Niemowlęta z SMA typu 1 są zwykle wiotkie, nieruchome, a nawet wiotkie. Ich języki poruszają się jak robaki i nie mogą trzymać głowy w pozycji siedzącej.
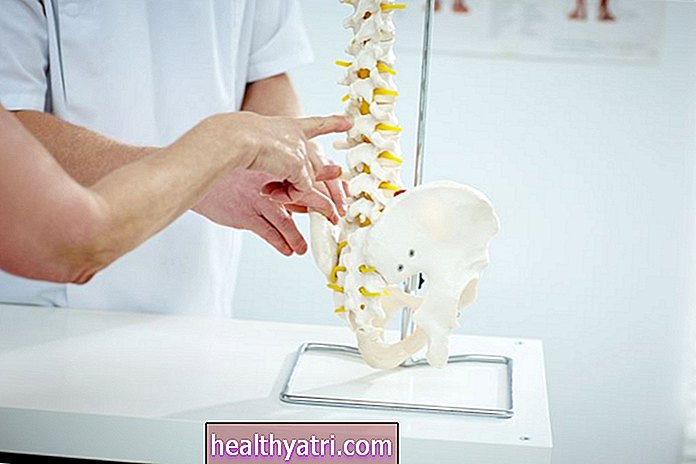
Mogą również mieć zauważalne deformacje, takie jak skolioza, i będą mieć osłabienie mięśni, szczególnie w mięśniach proksymalnych, które znajdują się blisko kręgosłupa.
Wpisz 2
SMA typu 2, zwana również pośrednią SMA, jest najpowszechniejszym typem SMA. Infekcja dróg oddechowych jest najczęstszą przyczyną śmierci w typie 2. Jednak dzieci z typem 2 mogą mieć normalną długość życia.
SMA typu 2 rozpoczyna się między 6 a 18 miesiącem życia lub po zademonstrowaniu przez dziecko, że potrafi siedzieć bez podparcia (po ułożeniu w pozycji siedzącej). Objawy typu 2 obejmują deformację, opóźnienie ruchowe, powiększenie mięśni łydek i drżenie palców.
Najpierw osłabione są mięśnie bliższe, które leżą najbliżej kręgosłupa; nogi słabną przed ramionami. Dzieci z SMA typu 2 nigdy nie będą mogły chodzić bez pomocy. Dobra wiadomość jest taka, że dziecko z SMA najprawdopodobniej będzie w stanie wykonywać zadania rękami i rękami, takie jak gra na klawiaturze, karmienie itp.
Zaobserwowano, że dzieci z SMA typu 2 są bardzo inteligentne. Fizjoterapia, urządzenia wspomagające i elektryczne wózki inwalidzkie mogą znacznie przyczynić się do ich sensownego życia.
Dwa główne problemy związane z SMA typu 2 obejmują:
- Słabe mięśnie oddechowe powodujące infekcję
- Skolioza i / lub kifoza rozwijająca się z powodu słabych mięśni kręgosłupa
Typ 3 i 4
SMA typu 3, znany również jako łagodny SMA, rozpoczyna się po 18 miesiącach. Osoby z SMA typu 3 są zwykle zależne od urządzeń wspomagających i przez całe życie muszą stale monitorować, gdzie się znajdują, pod względem ryzyka związanego z oddychaniem i skrzywieniem kręgosłupa. Mają tendencję do zaprzestania chodzenia na jakiś czas w swoim życiu. Kiedy przestają chodzić, wahają się między okresem dojrzewania a 40. rokiem życia.
Podczas gdy dzieci z SMA typu 3 mogą się poruszać i chodzić, dochodzi do osłabienia mięśni i zaniku mięśni proksymalnych, czyli najbliższych kręgosłupa.
Istnieje czwarty typ SMA, SMA początku dorosłych. Typ 4 zwykle pojawia się, gdy dana osoba ma 30 lat. Jak można się domyślić, SMA typu 4 jest najłagodniejszą postacią w kontinuum ciężkości tej choroby. Objawy typu 4 są bardzo podobne do objawów typu 3.
Przyczyny
SMA to zaburzenie genetyczne, w którym gen kodujący białko mięśniowe zwane SMN (neuron motoryczny przetrwania) jest uszkodzony. Nieprawidłowe działanie białka SMN prowadzi do problemów obserwowanych w SMA.
SMA jest dziedziczona recesywnie. Oznacza to, że aby mogło dojść do SMA, dziecko musi odziedziczyć wadliwy gen od obojga rodziców, a zatem oboje rodzice muszą być nosicielami wadliwego genu. Szacuje się, że około jedna na 40 osób jest nosicielem tego genu. Jeśli oboje rodzice są nosicielami, istnieje jedna czwarta szansy, że urodzone przez nich dziecko będzie miało SMA.
U niektórych osób z SMA inne geny mogą częściowo kompensować ten, który wytwarza wadliwe białka SMN. W rezultacie ciężkość SMA jest nieco zmienna w zależności od osoby.
Diagnoza
Pierwszym krokiem do postawienia diagnozy jest zauważenie przez rodziców lub opiekunów objawów SMA u ich dziecka, opisanych w tym artykule. Lekarz powinien przeprowadzić szczegółową historię medyczną dziecka, w tym wywiad rodzinny i badanie przedmiotowe.
Istnieje kilka rodzajów testów używanych do diagnozowania SMA:
- Badania krwi
- Biopsja mięśni
- Testy genetyczne
- EMG
Generowanych jest wiele problemów związanych z badaniami na SMA u dzieci, a także testowaniem rodziców pod kątem statusu nosiciela. W 1997 r. Na rynku pojawił się test DNA, zwany ilościowym testem PCR dla genu SMN1, aby pomóc rodzicom określić, czy są nosicielami zmutowanego genu powodującego SMA.
Test przeprowadza się poprzez pobranie próbki krwi. Badanie populacji ogólnej jest zbyt trudne, dlatego jest zarezerwowane dla tych, którzy mieli w rodzinie osoby z SMA. Badanie jest możliwe w okresie prenatalnym poprzez amniocentezy lub próbki kosmówki kosmówki.
Bardzo dobrze / Lara AntalLeczenie
Leczenie SMA koncentruje się na podtrzymywaniu życia, zachęcaniu do niezależności i / lub poprawie jakości życia pacjenta. Przykłady metod opieki i leczenia obejmują:
- Fizykoterapia
- Korzystanie z urządzeń wspomagających, takich jak wózki inwalidzkie, maszyny do oddychania i rurki do karmienia. (Istnieje wiele urządzeń wspomagających SMA. Najlepiej omówić to z zespołem terapeutycznym).
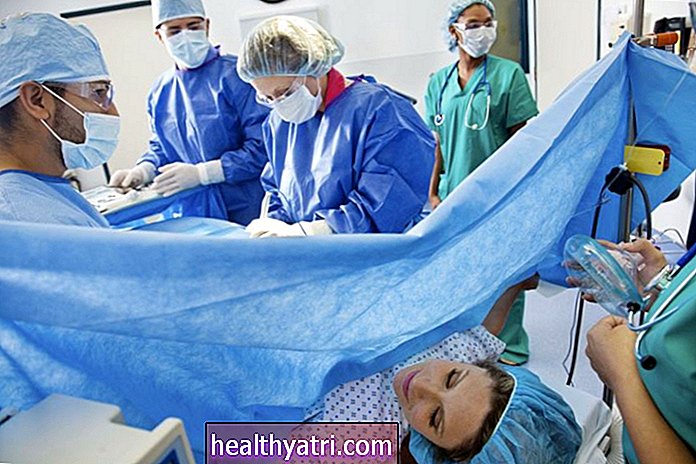
- Chirurgia deformacji kręgosłupa
Lekarze zalecają rodzinom współpracę z zespołem opieki zdrowotnej w sposób multidyscyplinarny. Pacjent z SMA powinien być często poddawany ocenie medycznej w ciągu jej życia. Poradnictwo genetyczne dla członków rodziny jest bardzo ważne.
Nie należy unikać aktywności, ale raczej stosować ją w taki sposób, aby zapobiec deformacji, przykurczom i sztywności oraz zachować zakres ruchu i elastyczność. Dlatego nie należy tego robić aż do wyczerpania. Dobre odżywianie pozwoli pacjentowi również wykorzystać swoje mięśnie.
Rdzeniowy zanik mięśni (SMA) - rzadki rodzaj osłabienia mięśni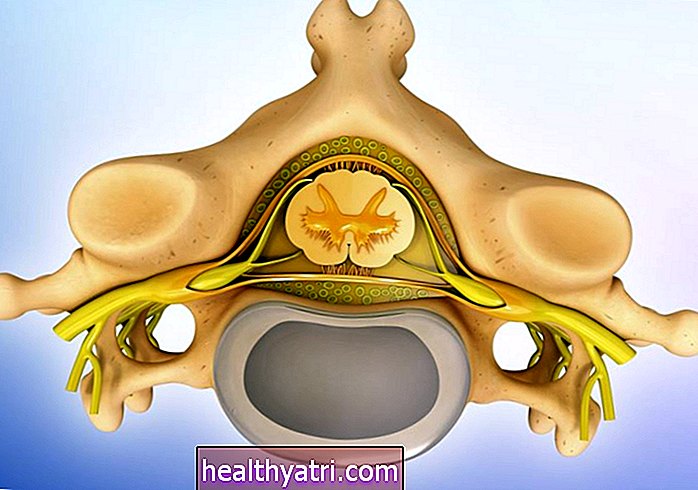















-is-treated.jpg)