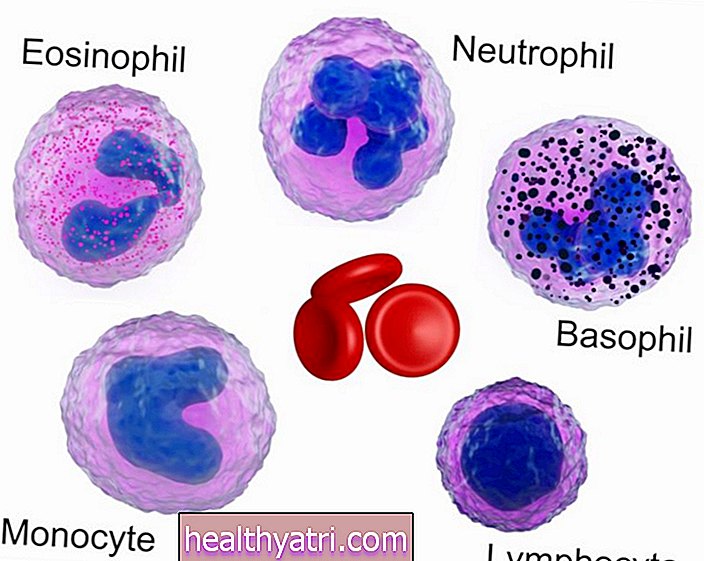
Talasemia beta jest dziedziczną chorobą krwi wywołaną mutacjami genów, w których organizm wytwarza zbyt mało hemoglobiny - białka w krwinkach czerwonych, które przenosi tlen w całym organizmie. W tym stanie niszczone są również czerwone krwinki. Z biegiem czasu niewystarczająca hemoglobina może prowadzić do anemii (niskiej liczby czerwonych krwinek lub RBC) i innych powikłań. Osoby z najcięższą postacią zaburzenia, poważną postacią talasemii beta lub anemią Cooleya, są zazwyczaj leczone regularnymi transfuzjami krwi; osoby z mniejszym typem, beta talasemią pośrednią, mogą rzadziej wymagać transfuzji.
Violka08 / Getty ImagesRodzaje
Istnieją trzy rodzaje talasemii beta.
- Cecha talasemii beta, w której osoba jest nosicielem genu zaburzenia, ale nie ma objawów. Czasami nazywa się to talasemią mniejszą.
- Beta talasemia pośrednia, stosunkowo łagodna postać zaburzenia
- Poważna postać talasemii beta lub anemia Cooleya, która jest najcięższą postacią
Talasemia beta występuje najczęściej u osób pochodzenia śródziemnomorskiego, azjatyckiego, indyjskiego i afrykańskiego.
Objawy
Objawy talasemii beta zależą od rodzaju i ciężkości zaburzenia. Osoby z mniejszą beta talasemią mogą mieć bardzo łagodną anemię, ale zwykle nie mają zauważalnych objawów. Niektórzy mogą nawet nie wiedzieć, że są nosicielami zmienionego genu.
Chociaż dwa zmutowane geny HBB są obecne po urodzeniu, dzieci urodzone z pośrednią talasemią beta lub anemią Cooleya mogą nie mieć objawów przed upływem 3 do 6 miesięcy; u niektórych dzieci z talasemią beta objawy występują dopiero w wieku 2 lat.
Niezależnie od tego, kiedy u dziecka z talasemią beta pojawiają się objawy, prawdopodobnie wystąpią u niego następujące objawy:
- Niedokrwistość
- Zmęczenie
- Słabość
- Duszność
- Żółtawa lub blada skóra
- Powolny wzrost
- Ciemny mocz
- Słaby apetyt
- Grymaszenie
W przypadku osób z niedokrwistością Cooleya objawy mogą się rozwijać i mogą wystąpić komplikacje, takie jak:
- Obrzęk brzucha
- Deformacje kości lub złamania kości spowodowane zmianami w szpiku kostnym, w którym wytwarzane są czerwone krwinki
- Powiększenie śledziony, stan, w którym śledziona, która filtruje czerwone krwinki, jest przepracowana i ulega powiększeniu
- Infekcje
Diagnoza
Chociaż przez większość czasu talasemia major jest identyfikowana na ekranie noworodka, osoby z talasemią pośrednią mogą zostać zidentyfikowane dopiero po latach. Osoby te są na ogół identyfikowane na podstawie rutynowej pełnej morfologii krwi (CBC). CBC ujawni łagodną do umiarkowanej anemię z bardzo małymi czerwonymi krwinkami. Można to pomylić z niedokrwistością z niedoboru żelaza.
Diagnozę potwierdza profil hemoglobiny (zwany także elektroforezą). W pośrednich mediach i cechach talasemii beta test ten ujawnia podwyższenie poziomu hemoglobiny A2 (druga postać hemoglobiny dorosłej), a czasami F (płód). Alfa talasemia pośrednia jest ogólnie nazywana chorobą hemoglobiny H, ponieważ jest to dominująca hemoglobina widoczna w profilu.
Talasemię beta można zdiagnozować za pomocą różnych badań krwi. Umiarkowana i ciężka beta talasemia jest zwykle diagnozowana we wczesnym dzieciństwie lub w wieku 2 lat, ponieważ objawy przedmiotowe i podmiotowe, takie jak niedokrwistość, zwykle pojawiają się bardzo wcześnie. Jeśli twoi rodzice cierpią na tę chorobę, być może zostałeś przebadany i zdiagnozowany przy urodzeniu lub w wieku niemowlęcym, zwłaszcza jeśli wykazywałeś niedokrwistość w bardzo młodym wieku.
Różne testy stosowane do diagnozowania talasemii beta obejmują:
- Badanie przesiewowe noworodka, które jest testowane rutynowo u większości niemowląt po urodzeniu, w celu wykrycia wszelkich chorób dziedzicznych. Może to wykryć talasemię beta lub przynajmniej czerwoną charakterystykę zaburzenia we krwi w celu dalszego monitorowania przez hematologa. Badanie noworodka polega na pobraniu małej próbki krwi i wysłaniu jej do laboratorium w celu oceny.
- Pełna morfologia krwi (CBC), która mierzy hemoglobinę oraz ilość i rozmiar czerwonych krwinek. CBC ujawni również łagodną do umiarkowanej niedokrwistość z bardzo małymi czerwonymi krwinkami, które są typowe dla talasemii beta. (Czasami jednak diagnozę można pomylić z niedokrwistością z niedoboru żelaza)
Osoby z łagodniejszymi postaciami talasemii mogą zostać zdiagnozowane po rutynowym badaniu krwi wykazującym niedokrwistość. Lekarze mogą również podejrzewać i badać beta-talasemię u osób pochodzenia afrykańskiego, śródziemnomorskiego lub wschodnioazjatyckiego, u których występuje niedokrwistość.
- Liczba retikulocytów, która określa stan szpiku kostnego i często jest częścią badania w kierunku anemii, może wskazywać, że szpik kostny nie wytwarza wystarczającej liczby czerwonych krwinek.
- Sprawdzenie poziomu żelaza we krwi pokaże, czy przyczyną niedokrwistości jest niedobór żelaza czy talasemia.
Ponieważ talasemia beta jest chorobą genetyczną przenoszoną z rodzica na dziecko, kilka innych testów, które mogą pomóc przewidzieć obecność genu i możliwe nasilenie talasemii beta:
- Elektroforeza hemoglobiny, która jest badaniem krwi dla rodziców, mającym na celu wykrycie różnych nieprawidłowości genetycznych w budowie hemoglobiny, może wykazać poziom hemoglobiny.
- Badania prenatalne mogą ujawnić nasilenie talasemii beta w przypadkach, gdy rodzice są nosicielami tego genu. Mogą to być: pobranie próbek kosmówki kosmówkowej około 11. tygodnia ciąży, usunięcie niewielkiego kawałka łożyska do oceny; oraz amniopunkcja, która jest wykonywana około 16 tygodnia ciąży i obejmuje badanie próbki płynu otaczającego płód.
Leczenie
Standardowe metody leczenia osób z umiarkowaną lub ciężką talasemią beta to transfuzje krwi, terapia chelatująca żelazo i suplementy kwasu foliowego:
- Transfuzje krwi: Transfuzja czerwonych krwinek może zrekompensować niedobór i zapewnić normalny poziom hemoglobiny. Osoby z ciężką postacią talasemii beta często wymagają transfuzji co dwa lub cztery tygodnie przez całe życie; osoby z beta talasemią pośrednią mogą wymagać jedynie sporadycznych transfuzji, w okresach choroby lub dojrzewania.
- Terapia chelatująca żelazo: pomaga organizmowi usunąć nadmiar i potencjalnie szkodliwe ilości żelaza z krwi - co może wynikać z wielu transfuzji. Chelatację żelaza podaje się doustnie lub w postaci wlewu podskórnego.
- Splenektomia: stosowanie tej procedury spadło w ostatnich latach, ale czasami rozważa się usunięcie śledziony, gdy terapia chelatująca nie działa.
- Suplementy kwasu foliowego: kwas foliowy jest witaminą z grupy B, która pomaga w budowie zdrowych czerwonych krwinek. Twój lekarz może zalecić suplementy kwasu foliowego oprócz transfuzji krwi i chelatacji żelaza.
- Przeszczep szpiku kostnego i komórek macierzystych: Przeszczep komórek macierzystych krwi i szpiku zastępuje wadliwe komórki macierzyste zdrowymi pochodzącymi od innej osoby (dawcy). Komórki macierzyste to komórki szpiku kostnego, które wytwarzają czerwone krwinki. Chociaż może to być możliwe lekarstwo na talasemię beta, tylko niewielka liczba osób z ciężką talasemią może znaleźć dobrego dawcę do tej ryzykownej procedury.
Osoby, które są nosicielami talasemii beta lub u których zidentyfikowano tę cechę, ale mają łagodne objawy lub nie mają ich wcale, zwykle wymagają bardzo niewielkiego leczenia.
Słowo od Verywell
Jeśli Ty lub członkowie Twojej rodziny masz beta talasemię lub gen beta talasemii, warto porozmawiać z lekarzem, jeśli myślisz o posiadaniu dzieci. Doradca genetyczny może pomóc określić ryzyko przeniesienia choroby na twoje potomstwo. Jeśli spodziewasz się dziecka, a Ty i Twój partner jesteście nosicielami talasemii, warto rozważyć wykonanie badań prenatalnych. Chociaż testy te wprawdzie wydają się przerażające i złowrogie, w rzeczywistości złagodzą Twój niepokój i pomogą Ci być przygotowanym, niezależnie od wyniku.

-count.jpg)








.jpg)